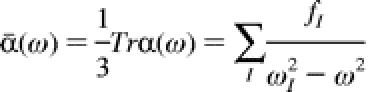-
Photodegradation of Sulfonylurea Molecules: Analytical and Theoretical DFT Studies
C. Corminboeuf, F. Carnal, J. Weber, J.-M. Chovelon and H. Chermette
Journal of Physical Chemistry A, 107 (47) (2003), p10032-10038


DOI:10.1021/jp035776b | unige:3703 | Abstract | Article HTML | Article PDF
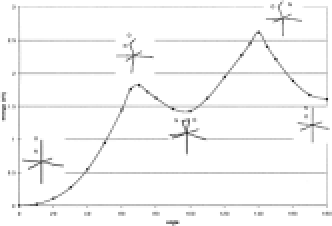
The use and the number of sulfonylurea herbicides have increased since the early 1980s. A good understanding of their degradation is of ecological importance, since environmental pollutants can be issued from them. It is claimed that microbial degradation and chemical hydrolysis present the main degradation pathways but photodegradation cannot be neglected. Time-dependent density functional theory has been used to help in the elucidation of the photochemical behavior of sulfonylureas.